An Introduction to the Biopharmaceutical Industry
David Gemmell looks at key unit operations, and why chemical engineers are heavily involved in their design, troubleshooting or optimisation
TO DATE, the Covid-19 pandemic has taken the lives of over 5.7m1 people and impacted global trade, travel, and the livelihoods of billions. It also sparked a vaccine arms race with worldwide political consequences. These events have brought biopharmaceuticals to the forefront of public discussions, but what are biopharmaceutical medicines and how do chemical engineers play their part in this cutting-edge industry?
This article provides an overview of the biopharma industry, highlights the role of chemical engineers, and sets the stage for subsequent articles that will explore additional aspects of the industry leading the charge in the battle against the pandemic. The series will provide a behind-the-scenes look into the products, key unit operations, required skillsets, and the future of the industry.
The basics of biopharmaceuticals
Biopharmaceuticals, broadly speaking, are therapeutic products – often proteins – produced using living systems, such as bacteria, yeast, or animal cells. The cells are first manipulated to introduce specific genetic material which directs them to use their own internal organelles and cellular machinery to manufacture a complex drug product.
In all living cells, proteins are of crucial importance for facilitating the functions necessary to sustain life. These large complex molecules catalyse chemical reactions, facilitate transportation of material, target and destroy foreign invaders and act as signalling devices – telling your organs what to do, and when.
The first human protein to be produced through biotechnological means was approved by the United States Food & Drug Administration (USFDA) in 1982, to the relief of diabetics everywhere2. That product was insulin, or more specifically a biosynthetically-derived human insulin distinct from the animal-derived variants that preceded it. Human genes which code for the insulin molecule were integrated into a ring of DNA called a plasmid. This plasmid was then inserted into E. coli bacteria which then synthesised the insulin as it would any other protein it naturally produces. This breakthrough of producing a “human” protein in a bacterial cell was the humble beginning of what has become an industry that has created hundreds of life-saving and life-enhancing treatments. These treatments include blood plasma products, antibiotics, recombinant proteins, monoclonal antibodies, antibody drug conjugates, and various types of vaccines.
A “human” protein in a bacterial cell was the beginning of an industry that has created hundreds of life-saving treatments
One of the most successful products engineered by the biopharmaceutical industry are monoclonal antibodies – mAbs. They are highly selective antibodies which bind to a specific target – called an antigen – in the body. This binding can facilitate destruction of the target, interfere with its function, activate it or lead to another outcome, based on the objective of the therapeutic.
In 1986 the first mAb was approved by the USFDA. This drug was designed to help patients prevent their bodies rejecting kidney organ transplants3 and while it was somewhat effective, major improvements in molecular specificity and therapeutic efficacy have been made for these therapeutic modalities.
Current-generation biologics such as mAbs, and other recombinant proteins offer a highly effective, precise medication for significant diseases ranging from cancers to rare genetic disorders, autoimmune diseases and more. Let’s take a look at how these powerful drugs are manufactured.
mAbs are generally made using Chinese hamster ovary (CHO) cells, mammalian cells cloned originally from hamster ovary cells decades ago. A gene coding for the therapeutic protein is spliced into the CHO cell by means of recombinant DNA technology. Cellular machinery (RNA Polymerase) transcribes this DNA into messenger RNA (mRNA) which is then translated into proteins – the mAb – which are secreted from the cell. These proteins can then be separated from the solution in which the CHO cells were grown and purified.
A biopharmaceutical manufacturing process consists of two parts, upstream production and downstream purification. The upstream phase involves the selection of appropriate cells (most often the CHO cells described above), cell culture media and feeds (specific solutions of amino acids, vitamins, sugars, and other components that the cells will use as a source of nutrition) and means of containing these cells within a sterile boundary while providing the optimum conditions necessary to sustain them and allow them to multiply and produce the desired protein product.
Bioreactors are vessels designed to house the cells, keeping them supplied with the necessary nutrients and maintaining the most efficient environmental conditions to maximise protein yield. Bioreactors are typically designed to mimic the conditions cells would experience in a living body, specifically an operating temperature of 37oC, along with specific pH, dissolved oxygen and CO2 levels. The bioreactor volume at the end of a production run will contain waste materials as well as the protein of interest. These process impurities will include whole cells, dead cells, cellular debris, host cell DNA, host cell protein and possibly other process related contaminants. The downstream phase is designed to separate the process impurities from the molecule of interest.
Chemical engineers are heavily involved in the design, troubleshooting or optimisation of all upstream and downstream unit operations. Throughout the entire process, many classic aspects of chemical engineering are applied: mixing of non-Newtonian solutions, heat transfer, mass transfer, sterile or aseptic material transfer, design of cleaning systems, sterilisation systems, pressure vessels and process control. The key unit operations for a mAb process are shown in Figure 1.
Drug manufacturers have developed a templated approach over the last two decades which has helped to significantly reduce production costs, allowing the production of different products in different facilities with only minor process modifications. This allows drug manufacturers to construct production facilities which can handle multiple products without significant new investment. While the mAb process can be templated, not all biological therapeutics lend themselves well to a templated approach due to their inherent variability.
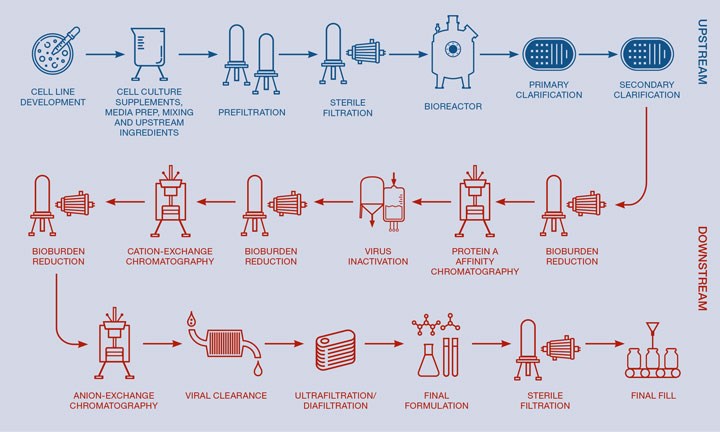
Key unit operations in upstream production
In addition to cell line/cell culture scaleup and media development, the key upstream unit operations are the sterilisation, by heat or filtration, of all components that will enter the bioreactor, the growth of cells and subsequent protein expression within the bioreactor, and the harvesting and initial purification step which is referred to as clarification.
Maintaining sterility is key to an effective biomanufacturing process as the conditions within the bioreactor will facilitate effective growth of cells, whether they are the intended cell or an adventitious agent which has infiltrated the sterile system. These harmful agents could be bacteria, mycoplasma, fungi, or viruses. There are many techniques to ensure that sterility is maintained but a key step in all biopharmaceutical processes is sterile filtration.
Sterile filtration is typically achieved with devices made from either polyethersulphone (PES) or polyvinylidene fluoride (PVDF) membranes. These membranes have been designed to have hydrophilic surface chemistry and microscopic pores which allow protein molecules to permeate through while stopping larger contaminants like bacteria. Sterile filters typically have a 0.22 µm pore size, which will retain the smallest bacterium known B.dimunuta, to a high level reduction. However 0.1 µm filters are available to remove myxoplasma which are smaller than bacteria. Combinations of different pore sizes can be used to maximise the capacity of the filters, while maintaining sterility assurance. These filters can also be used to minimise bioburden – bacteria or biologic contamination – at points in the process where true sterility is not critical. Sterile and viral filtration filters operate in normal flow filtration (NFF) mode, with the flow of liquid directed through the filter membrane.
The clarification step, which comes after the completion of the cell culture stage, begins the separation and purification of the therapeutic protein from host cells, host cell debris, lipids, host cell DNA and other contaminants. There are many different strategies for clarification, and these must be assessed by biomanufacturing/biochemical engineers when designing the manufacturing process and facility.
Manufacturing scale and batch size will play a part in this decision-making process. Many companies who operate large stainless-steel bioreactors with capacities of 10-20,000 L typically elect to use a centrifugation system to begin separation of the bioreactor output. A common strategy is to utilise centrifugation followed by depth filtration. However, there are other options such as tangential flow filtration (TFF), chemical flocculation within the bioreactor, or any combination of the above.
As industry increases the ability to get higher titres of product from the same volume of cells in a bioreactor, smaller more flexible processes are being routinely used. Selecting a process which uses single-use depth filtration devices without centrifugation can allow for a significant reduction in capital costs and a decrease in process cycle time as the filters are discarded once the batch is processed and fresh ones installed for the next run. The mitigation of lengthy cycle times for cleaning and sterilisation of stainless-steel hardware (and the mitigation of the hardware purchase cost itself) can lead to significant cost reductions which can be transferred to an overall lower cost of goods.
Depth filters use larger open pore filtration media from both natural sources such as diatomaceous earth media, cellulose fibres, or synthetic media, arrayed in a thick sheet. The open fibrous strands of these media types are often given an electrostatic charge profile. Particle removal occurs either by size exclusion, adsorption/charge interaction and impaction.
Once clarification is complete and the physically larger process impurities such as cells are removed the next phase can begin. Clarification is typically followed by a sterilising/bioburden control filter. This will allow for a hold stage in transferring to the downstream purification or to maintain low contamination levels prior to the start of the next steps
Key unit operations in downstream purification
The true workhorse in any mAb purification process is the affinity chromatography column. Affinity chromatography consists of a column containing microporous resin media (the solid phase), which has a specific ligand bound to the resin. This ligand is a protein which has been isolated from Staphylococcus aureus bacteria4 either directly or produced by recombinant means, and binds to the Fc region of mAbs. The column is equilibrated using specific buffer solution (mobile phase) which provides the optimal pH and conductivity conditions for the ligand to express its biochemical properties, preparing it for the product-containing feed stream to flow through the column.
The majority of process impurities in the product stream will not bind to the affinity ligand, allowing the isolation of the mAb, and facilitates a significant reduction in process volume and smaller subsequent processing steps. Once the product has been loaded onto the column, the elution cycle will begin. A buffer with a lower pH or conductivity will change the interaction between the ligand and the adsorbed protein. Altering the biochemical characteristics, results in the mAb no longer binding and its desorption (elution) from the column. The output stream from the column can be selectively collected with specific volumes known as fractions, and are then separated, collected, or sent to waste.
Affinity chromatography can provide upwards of 95% protein purification in a single unit operation. However, not all impurities will be removed. Host cell proteins or host cell DNA which have become loosely attached to the mAb product will be bound to the column and could be co-purified during mAb elution from the column. Further purification steps which exploit different biochemical characteristics of the target mAb are necessary to attain strict product purity specifications.
The affinity capture step will typically elute at low pH (typically pH 3), and the product-containing fraction is held at these conditions as part of the viral safety strategy. Viral inactivation is typically done chemically, either by solvent/detergent treatment or by a low pH hold for up to one hour. This step is effective on larger enveloped viruses, such as those which may have contaminated the upstream process or have been produced by cells that were already infected.
The product stream is further purified using other forms of chromatography. Ion exchange chromatography (IEX) is a common choice for what are known as polishing steps. A cation exchange column run in bind/elute mode will remove species with a different charge from the product and a subsequent flow through anion exchange column will remove negatively charged impurities, allowing the positively charged mAb product to flow through and be collected with purities of 99% and above.
The delicate biological product is now nearing the end of its purification process. Regulators mandate that orthogonal viral clearance steps are incorporated into the purification process, meaning that the steps used to clear virus should operate by different means of separation or inactivation. Chromatography can remove certain viruses based on their affinity for the column media, low pH/detergent holds provide inactivation, and the final step is typically completed by size exclusion.
Virus filtration is performed by devices with membrane pore sizes in the nanometre scale. These systems can be challenging to develop as the protein products may have only small differences in physical size compared to the smaller viruses in the parvovirus family.
The drug product is then typically concentrated to a pre-determined setpoint and the buffer which has been carrying it through these steps may be adjusted or replaced with a new buffer to keep the drug product stable for an extended duration.
Tangential flow filtration (TFF) systems are well suited to this application. TFF systems use a size exclusion process with flow directed tangentially over the filter, as opposed to directly through it. TFF can be used to separate a product into the permeate (filtrate) when the protein is smaller than the
molecular weight cut-off (MWCO) of the TFF membrane or collect the product on the retentate side and concentrate it, when the protein is larger than the MWCO of the membrane.
Ultrafiltration/Diafiltration (UF/DF) steps using TFF allow the mAb product to be concentrated on the retentate side of the filter while buffer salts and small impurities are allowed to permeate through. Allowing new buffer to enter the retentate vessel at the same rate which old buffer is leaving through the permeate line allows protein concentration to be maintained while a more suitable solution, such as the final formulation buffer, is exchanged in.
Once the desired protein concentration is achieved, and the final formulation of the bulk drug product is completed, the solution is ready to be sterile filtered and filled into final packaging.
Chemical engineers are involved everywhere in the manufacturing process, from designing the manufacturing facility, to designing each unit operation, the supporting utility systems providing purified steam or water to be used for equipment sterilisation, and the hydration of buffers/powdered cell culture media. Engineers also undertake mass and energy balances to assess opportunities for yield improvement, process optimisation or loss prevention. Careful scheduling is almost always required to ensure that the upstream bioreactor which may have a cycle time of several days to multiple weeks is balanced with the downstream purification train to maintain high equipment utilisation. Larger facilities may have several bioreactors and multiple purification trains.
Knowledge of the rigorous hygienic design codes defined in the American Society of Mechanical Engineers Bioprocessing Equipment design guide (ASME BPE) and the European equivalent (European Hygienic Equipment Design Group-EHEDG) is critical to ensure that systems which contact the drug product are designed and maintained to achieve the highest standards of cleanliness. Biological products are delicate and typically shear and heat sensitive, so standard equipment selections or processing techniques may not be feasible.
Even the most inexpensive drugs can still be worth thousands of dollars per millilitre of pure drug product; as such, process optimisation is critical for companies seeking to lower their cost of goods.


Future outlook
Biopharmaceuticals are expensive products to manufacture. Investment in the industry has increased even further in the wake of the Covid-19 pandemic, with significant funding being poured into the UK to further develop a manufacturing base. In Ireland, biopharmaceuticals is a large industry accounting for up to €15bn (US$16bn) in corporation tax with approximately 120 foreign companies and nine out of the top ten largest drug producers having significant manufacturing sites.
It is unlikely that the UK would be able to successfully pursue this scale of manufacturing; however, as larger, more capital intensive stainless-steel based manufacturing processes are replaced with smaller, single-use plastic systems, the cost to design and build a new facility is decreasing.
Industry expects this trend to continue with the development of smaller more flexible multi-product facilities moving away from traditional batch manufacturing strategy to a more continuous model adopted by almost all other industries. While technological and regulatory challenges remain, with time these will be overcome by scientists and engineers working together.
Increasingly sophisticated manufacturing strategies are on the horizon and include applications like viral gene therapy. Focusing on DNA/mRNA manufacturing, facilities of the future may be revolutionised so that they only produce a specific piece of mRNA that will be translated into a protein drug product, such as a mAb, by ribosomes in your own body. Additionally, the target mRNA molecule can be easily changed from batch to batch.
This level of unprecedented flexibility could possibly alter the biopharmaceutical landscape forever, allowing us to inch ever closer to a personalised medicine approach. Once a pipedream, the thought of testing a patient and creating a bespoke therapeutic swiftly and cost effectively is now closer than ever.
Subsequent articles in our biopharmaceutical series will discuss some of the exciting trends being observed in industry form novel modalities like mRNA to the challenges with continuous bioprocessing. I hope these discussions provide an insight into a truly fascinating industry.
Globally, biological systems are being researched for many different applications, from biocomputing, to enzymes to breakdown plastic waste and biofuels, to water purification. While there are many hurdles and uncertainties to overcome, one thing is clear. The future is bright, it is almost certainly biological, and there are ample opportunities for chemical engineers.
References
1. WHO Coronavirus (Covid-19) Dashboard, 2022.
2. Quianzon, C and Cheikh, I, “History of insulin”, J Community Hosp Intern Med Perspect 2012, 2, (2), 10.3402/jchimp.v2i2.18701
3. Liu, JKH, “The history of monoclonal antibody development - Progress, remaining challenges and future innovations”, Ann Med Surg (Lond) 2014, 3, (4), 113-116.
4. Graille, M et al “Crystal structure of a Staphylococcus aureus protein A domain complexed with the Fab fragment of a human IgM antibody: structural basis for recognition of B-cell receptors and superantigen activity”, Proceedings of the National Academy of Sciences of the United States of America 2000, 97, (10), 5399-5404.
Acknowledgments: Thanks to Michael Burns, Paul Beckett, Stuart Rolfe and Laurence Carudel for their assistance in writing this article.
This is the first in an ongoing series of articles on how chemical engineers contribute to the biopharmaceuticals industry. To read the full series as it develops, visit: https://www.thechemicalengineer.com/tags/chemical-engineers-and-the-biopharmaceuticals-industry/
Recent Editions
Catch up on the latest news, views and jobs from The Chemical Engineer. Below are the four latest issues. View a wider selection of the archive from within the Magazine section of this site.



