Viral Safety Challenges in Bioprocessing
David Gemmell explains how the industry keeps viruses and bacteria at bay in manufacturing
THE penultimate article in our biopharmaceutical manufacturing series discusses the challenges in ensuring viral safety of therapeutic products. You will learn about the different technologies and strategies that ensure that viral and bacterial contamination risks are mitigated.
Out of the many activities that chemical engineers are tasked with in the biopharmaceutical industry, arguably none is more important than ensuring that the manufacturing process achieves the highest levels of product quality, and therefore patient safety on a routine, repeatable basis.
There are a variety of distinct types of biological contamination. Contamination from microorganisms such as fungi, mycoplasma, bacteria, and viruses can cause serious issues for manufacturers.
Many therapeutics are parenteral – meaning that they will be injected into the patient, bypassing many of the body’s mechanisms which have evolved to destroy a bacterial or viral threat, so the highest levels of quality are vitally important.
As discussed in the first article in this series (see TCE 970), sterilising filtration is used to control bioburden, removing bacteria and mycoplasma which may enter the process from the environment, operators, or raw materials. These technologies are used at multiple steps throughout both upstream cell culture processing and downstream purification.
The approach to viral safety depends on the final product being produced, and the position of the virus clearance unit operation in the overall manufacturing or purification process. Methods used upstream may not be appropriate downstream as the process fluids, volumes and manufacturing conditions are different. Similarly, methods designed for downstream virus risk mitigation are not appropriate for use in upstream processing.
Upstream viral safety
There are strict viral safety requirements and guidance documents available to direct manufacturers towards different approaches for mitigating the risk of viral contamination. However, the guidance is primarily focused on downstream purification to ensure the final drug product is not contaminated with virus.1 Upstream viral risk mitigation is primarily considered a business risk and, for now, there is no regulatory requirement to implement viral clearance technologies.
The multiple layers of control throughout biomanufacturing makes it highly unlikely an upstream virus contamination event would present a safety risk to patients. However, it has the potential to create significant issues which may indirectly put patients at risk. Viral contamination events can cause a facility to shut down while laborious root cause analysis is undertaken, and decontamination/remedial works occur. If the shutdown persists for a long duration, it could cause a shortage of drug products, creating challenges for the patients who need them.
This is not a hypothetical scenario. In 2009, a large manufacturing facility in the US suffered a significant Vesivirus 2117 contamination. The resulting shutdown caused a global shortage of two life-enhancing drugs and led to the US Food & Drug Administration (US FDA) levying fines which ran into hundreds of millions of dollars. The impact on the company was approximately US$1bn2 in lost revenue, cost of remedial/decontamination works, and FDA fines. Furthermore, the damage to the company reputation was so severe that it was swiftly bought out by a competitor.
The 2009 example is extreme but being able to implement measures to mitigate the cost of losing a batch is worth considering. Cell culture media and other consumables used in a bioreactor are incredibly expensive. Significant time is also required to make a batch: one manufacturing run may last between 5–30 days depending upon the product. For this reason, some companies implement upstream viral barrier technologies to provide extra mitigation against viral contamination. Implementing processes to systematically identify high-risk components and mitigate their threat should be strongly considered. Otherwise, there is a risk of losing significant sums of money as raw materials and time have been wasted if there is a contamination event.
Where do the risks come from?
In terms of viral contamination risks there are several potential vectors which could bring contamination into the bioreactor. Process operators/personnel are one source, as are the process utilities (air, water etc), the environment in the facility, the raw materials, or the cell line itself.
Endogenous viruses are present in most cell lines used to synthesise therapeutic proteins. Cell lines can be extensively screened for the presence of virus using a variety of sensitive methods to minimise this risk. More recently, there are cell lines available which have been engineered to be resistant or entirely unable to host specific viruses which might be an even safer option when compared with cell line screening.
Biopharmaceutical manufacturers operate rigorous hygienic and environmental control practices alongside cleaning regimes for non-single use systems. Therefore, typically, the biggest contamination risk will come from the raw materials used within the bioreactor.3
Historically, components of animal origin were used to provide the necessary nutrients cells needed to grow and be productive. Foetal Bovine Serum or Bovine Serum Albumin were commonly used. However, animal-derived components have high lot-to-lot variability and have a higher risk of containing viruses.
Plant-based components, while much safer than animal components, still retain a high viral risk. The industry is moving away from animal-derived components in favour of chemically-defined alternatives. These more appropriate materials are synthetic and have inherently lower risks.
Glucose is one of the most critical cellular components. It is typically sourced from sugar cane or beet producers. Sugar is a rodent attractant, and mammalian viruses such as Minute Virus of Mice (MVM) can be passed to the glucose material via rodent urine or faeces.
As many operators utilise Chinese Hamster Ovary (CHO) cells to produce the therapeutic protein drug product, any mammalian virus can be devastating. Glucose is often the highest-risk component for manufacturing processes that do not utilise raw materials of animal origin. As the vast majority of sugar is destined for the food and beverage industry, it is impossible for biopharmaceutical suppliers to persuade sugar producers/wholesalers to adopt the rigorous quality, pest and facility control practices that are commonplace at the biomanufacturing end-user facilities. This leads to a potential hazard which should be mitigated.
MVM, a parvovirus, is considered to be the worst-case virus contaminant for a mammalian expression system. These viruses are small in size (20-26 nm) and have no lipid envelope, resulting in high physico-chemical resistance and resistance to inactivation by low pH, detergent, or mid-temperature heating.
Parvoviruses are one of the panel of viruses manufacturers use for clearance evaluations; clearance data is expected as part of the regulatory approval process. This data must be generated to show that common worst-case viruses will be removed by the downstream purification process.
Upstream barrier technologies
In terms of upstream viral barrier technologies there are multiple options. Virus filtration of cell culture media can be performed with filters that have been designed to remove small and large viruses from typical cell culture media/feed streams. These filters are similar to downstream virus filters, but contain a membrane optimised for processing chemically-defined cell culture media.
Another technology which has been shown to be effective for treating cell culture media is High Temperature Short Time (HTST) pasteurisation. This technology is more prevalent in the food and beverage industry, but it has been used in biopharma since the early 90s.
Multiple companies operate their own in-house HTST processes to treat their bioreactor feeds. Designing and building an HTST process is typically expensive and extensive R&D efforts are required to determine which components can and cannot be effectively pasteurised.
As an alternative, some suppliers offer commercially-available HTST-treated raw materials/feeds. This provides a solution for biomanufacturers who either do not have the resources or do not desire to go through the capital-intensive design and build process. For companies with multiple manufacturing sites who wish to adopt a global viral risk mitigation strategy, having these components available from a trusted supplier is an attractive option. It is also useful for contract manufacturing organisations (CMOs) who do not have an HTST process in-house, but who want to mitigate viral contamination risk for their clients.
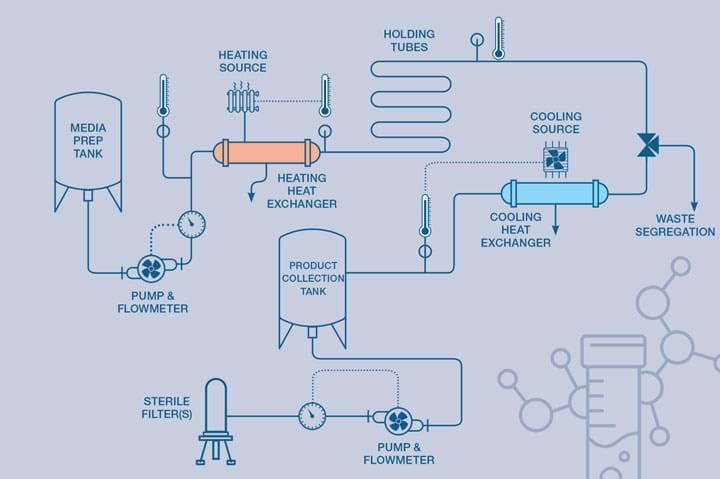
Chemical engineers must ensure that thermal inactivation systems like HTST can provide precise time and temperature control, making sure that the fastest fluid element is taken into consideration. Hygienic design practices are critical to ensure that the resulting system is fit for purpose.
High quality components and equipment are required to get optimal performance, and linking the system to a data management/historian system enables detailed quality records to be stored. HTST systems come in varying designs. More sophisticated systems monitor critical process parameters to ensure that critical quality attributes are met. Flow rate can be assessed against a known fluid pathway/volume so the residence time can be controlled. The inactivation temperature (typically 102–110oC) is most appropriately achieved with a heated water loop over the primary heating exchanger for enhanced temperature control and to mitigate the harsher effects of direct steam heating. Back pressure, conductivity, pH, turbidity, and log-mean temperature difference may also be monitored depending upon the application and raw materials being pasteurised.
Monitoring the log mean temperature difference across the heat exchangers can help determine if the exchangers are being fouled by over-heated products burning on to the tubes or plates. The HTST system is designed to ensure that all parameters are in specification while high quality water circulates. Then the liquid to be pasteurised is released into the system. The water and initial water-product interface is directed to drain and then valve switching enables the treated product to be collected. If one of the critical process parameters goes out of specification, the system should be designed to ensure that that material is automatically directed to waste and the collection vessel isolated to maintain product quality of the treated bulk. With careful design it is possible to achieve a 6-log reduction (99.9999%) of MVM using HTST.3
Alternative approaches to viral inactivation include ultraviolet (UV) light with a wavelength of 254 nm. There are vendors who provide lab-scale and larger-scale systems, but they are often more expensive, and more work is required to validate and utilise them. The inactivation is governed by the viral DNA (Deoxyribonucleic acid) strain, the consistency/depth of UV-C penetration of the raw material and the flow rate of the system. HTST is typically more widely adopted as it is much easier to validate time and temperature.
Components which are known to be of particularly high-risk such as Bovine Serum Albumin are often treated before use with gamma irradiation to inactivate virus.
Many single-use components (bags, filters, transfer assemblies or tubing) are supplied irradiated but this critical feature is creating a supply chase bottleneck. Cobalt-60 is one of the most common gamma emitters for the sterilisation of materials. However, this radioisotope is in short supply, and many suppliers are looking at alternative sterilisation methods, such X-ray sterilisation, to alleviate supply chain bottlenecks
Downstream viral safety
Demonstrating orthogonal mechanisms of viral clearance in downstream purification is a regulatory requirement. This approach should result in effective reduction for a broad panel of potential viral contaminants. Acceptable levels of viral clearance is typically achieved by a combination of chromatography, low pH inactivation and viral filtration steps (see TCE 970 for an overview of typical downstream purification unit operations).
Creating membranes that can separate protein products with approximate sizes of 5–15 nm and viruses with sizes of 20 nm and greater is challenging. Such a small difference in size creates challenges to produce a membrane that ensures robust viral removal while maintaining high process flux (filtration flow rate per filter area), capacity (volume of filtrate processed before filter blockage) and protein yield.
Making membranes
In general filtration, membranes are created by mixing polymers and solvents into a lacquer which corresponds to the specific membrane material desired. The lacquer is then cast onto a film which pulls the lacquer through a formation water bath. The resulting membrane is pulled through a series of baths to wash off any residual solvent. The membrane is collected and then subjected to chemical modification to alter the originally hydrophobic membrane surface and modify it to ensure hydrophilic properties. The membrane porosity and pore size is achieved by interactions between the lacquer and the formation bath. The modified membrane is then collected on rolls and cut to make flat sheets which can be layered onto/around support materials and feed channels before being housed in a cartridge, cassette, or capsule. At each step in this process, extensive controls need to be in place to assure quality specifications are met and virus filtration devices will reliably deliver the performance needed and expected by biomanufacturers.
An alternative to flat sheet/spiral wound devices is the use of hollow fibres which are gathered with endplates attached. This configuration can be imagined as a shell-and-tube heat exchanger tube bundle where the tubes meet endplates allowing liquid to enter/exit. The separation occurs as liquid flows through the fibre. Smaller particles pass through pores in the walls of the fibre, larger particles are retained.
In general, flat sheet devices are easier to scale up than hollow fibres, but both can be used given the correct process development work is undertaken. Chemical engineers are heavily involved in the R&D of the membrane casting/fibre spinning processes, the design and implementation of the casting equipment, and in the commissioning, validation, and operation of the process.
Different membrane manufacturing materials and techniques are utilised to make various products for the many process steps in the biopharmaceutical industry, from filtration devices to components in analytical devices such as pregnancy tests or rapid antigen tests. Outside of biopharma, the membranes will be used for the purification of water, food and beverages, air/gases (hydrophobic membranes) or other liquids.
New challenges – when virus is the product
The emerging sectors of cell, gene and viral therapy manufacturing are offering new opportunities for creation of different solutions to treat a wider variety of diseases and illnesses. Many companies are using viruses such as Adeno-Associated Virus (AAV) and Lentivirus as delivery vectors. These genetically-modified viruses contain the genetic sequences required to create a specific protein.
Although these offer new opportunities, they also create new challenges. As these viruses are the same size as the viral contaminants described previously, robust viral safety steps, such as size exclusion filtration, are harder to implement in downstream processing. In these instances, greater care and vigilance to select the highest quality raw materials should reduce the risk of introducing viral contaminations into the manufacturing process. Further consideration of upstream barrier technologies such as HTST-treated materials or viral barrier filtration may be more attractive to mitigate process viral safety risk.
Chemical engineers working with scientists are constantly developing new techniques to streamline and optimise the manufacturing of therapeutics. There are almost endless opportunities for chemical engineers to thrive in this industry, even without a biological processing background. There has never been a better time to join this industry and help to deliver life-changing technologies and medicines of the future.
References
1. FDA, Q5A Viral Safety Evaluation of Biotechnology Products Derived From Cell Lines of Human or Animal Origin, 1998.
2. Rockoff, JD, “Drug Manufacturing Mending After Questions of Quality”, Wall Street Journal, Vol 2010, New York.
3. Gemmell, DK et al “Efficacy of minute virus of mice (MVM) inactivation utilizing high temperature short time (HTST) pasteurization and suitability assessment of pasteurized, concentrated glucose feeds in Chinese hamster ovary (CHO) cell expression systems”, Eng Life Sci 2021, 21, (7), 502-513.
Acknowledgments: Michael Burns, Paul Beckett, Stuart Rolfe, Trish Greenhalgh and Laurence Carudel for assistance in writing this article.
This is the fourth in an ongoing series of articles on how chemical engineers contribute to the biopharmaceuticals industry. To read the full series as it develops, visit: https://www.thechemicalengineer.com/tags/chemical-engineers-and-the-biopharmaceuticals-industry/
Recent Editions
Catch up on the latest news, views and jobs from The Chemical Engineer. Below are the four latest issues. View a wider selection of the archive from within the Magazine section of this site.



